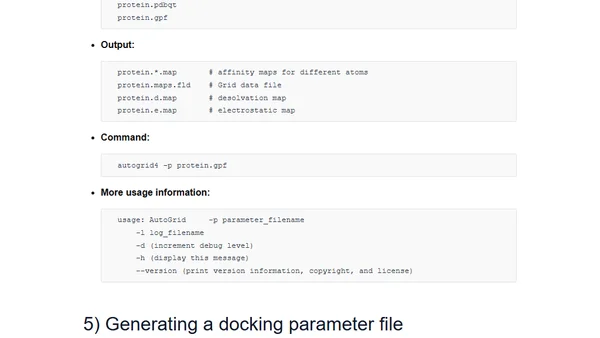
Molecular docking, estimating free energies of binding, and AutoDock's semi-empirical force field
Read OriginalThis article provides a detailed guide to molecular docking, explaining its goals of identifying ligand binding poses and estimating binding affinities. It focuses on the practical steps of using AutoDock 4.2's semi-empirical force field for binding energy estimation, covering protein and ligand preparation, grid generation, and docking execution. It also compares other scoring functions like AutoDock Vina, DrugScoreX, and LigScore.
Comments
No comments yet
Be the first to share your thoughts!
Browser Extension
Get instant access to AllDevBlogs from your browser
Top of the Week
1
The Beautiful Web
Jens Oliver Meiert
•
2 votes
2
When your coding agent doesn’t understand your project, you’ll get junk
Benjamin Cane
•
1 votes
3
LLM Use in the Python Source Code
Miguel Grinberg
•
1 votes
4
Wagon’s algorithm in Python
John D. Cook
•
1 votes
5
An example conversation with Claude Code
Dumm Zeuch
•
1 votes